新算法削减世界上最强大的显微镜的焦点 - 改善了Cryo-EM 3D分子结构地图
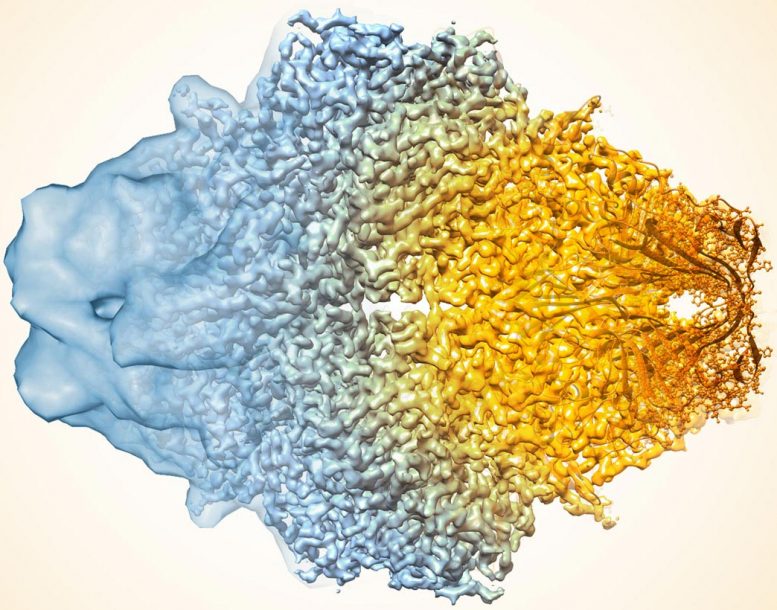
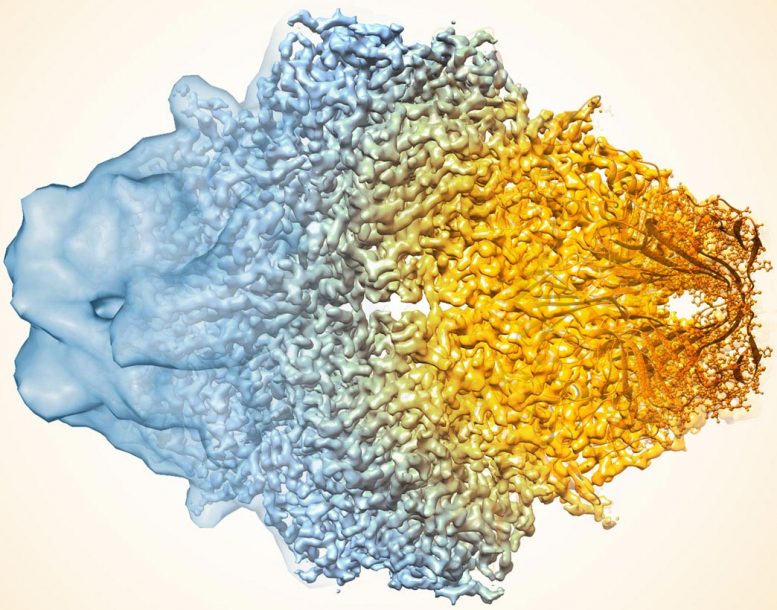
酶乳糖酶的复合图像近年来如何如何提高Cryo-Em的分辨率。较旧的图像到左侧,更近于右侧。
科学家开发一种改善冷冻电子显微镜的分辨率的技术。
我们都看到董事会电视节目中的那一刻,侦探正在审查颗粒状,低分辨率的安全镜头,斑点的一个感兴趣的人,并且不正当地向CSI技术人员提出“提升。”几个键盘稍后点击,而瞧 - 他们有一个完美的清晰的嫌疑人脸的图片。当然,这在现实世界中不起作用,因为互联网上的电影批评者和人们就像指出。
然而,现实的科学家最近开发了一个真实的“增强”工具:一种提高强大的显微镜的分辨率和准确性,这些方法用于揭示洞察生物学和医学的见解。
在一项研究中发表的自然方法中,由汤姆特拉米尔从新墨西哥财团领导的多机构团队,包括来自劳伦斯伯克利国家实验室(Berkeley Lab)的研究人员展示了新的计算机算法如何提高生成的3D分子结构地图的质量用冷冻电子显微镜(Cryo-EM)。
几十年来,通过占用许多显微镜图像和应用图像处理软件来生成的这些Cryo-EM地图 - 对于研究人员来说,寻求学习动物,植物,微生物和病毒功能的分子的关键工具。近年来,Cryo-EM技术已经推进了它可以为许多类型分子产生原子级分辨率的结构。然而,在某些情况下,即使是最复杂的Cryo-EM方法也仍然产生具有较低分辨率的地图,并且比梳理复合化学反应的细节所需的更高的不确定性。
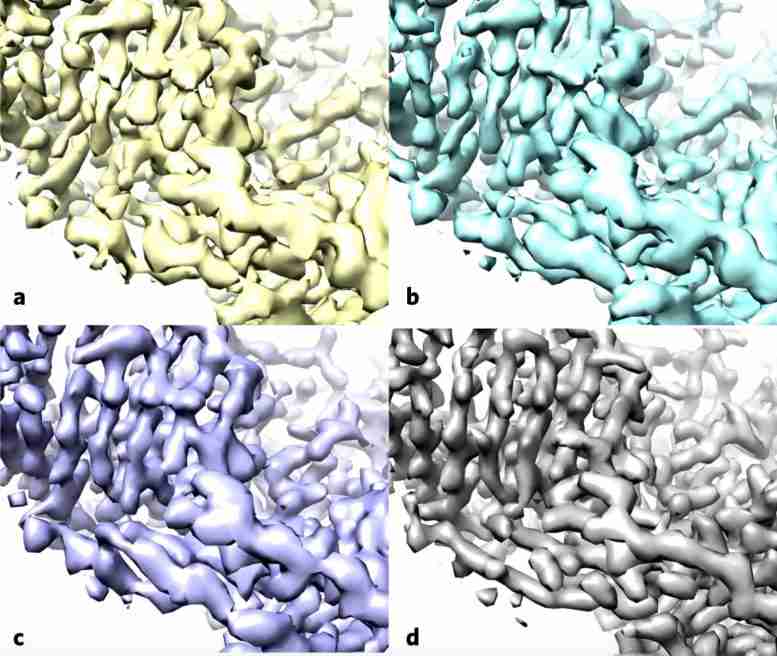
使用酶 -β 甘露出酶,也称为乳糖酶,作为测试用例,研究人员应用了标准方法(a),然后施加了没有(b)的改进算法,并用滤波器提高地图中的噪声均匀性(c) ,这两种地图与沉积的高分辨率蛋白质结构图(D)更类似于。
“在生物学中,我们通过了解分子的结构来获得这么多,”伯克利实验室的分子生物物理学与综合生物影像师司总监Paul Adams表示。“我们通过该算法看到的改进将使研究人员更容易从电子丧光 - 显微镜数据中确定原子结构模型。这对于建模非常重要的生物分子,例如参与转录和翻译遗传密码的那些,这尤其如此,这通常仅在较低分辨率地图中看到,这是由于它们的大而复杂的多单元结构。“
该算法通过基于存在的分子看起来的现有知识以及如何最佳估计和去除显微镜数据中的噪声(不希望的和不相关的数据)来削减分子映射。以前用于改善X射线晶体学生成的结构图的方法,并且科学家们之前提出了在Cryo-EM之前的使用。但是,根据亚当斯的说法,没有人能够表现出它在Cryo-Em工作的明确证据,直到现在。
该团队 - 由来自新墨西哥州的科学家组成,来自新墨西哥州的科学家,洛杉矶阿拉莫国家实验室,剑道大学和伯克利实验室 - 首先将该算法应用于已知有3.1-angstrom的人蛋白质植物蛋白的公开地图分辨率(Angstrom等于10亿米的10亿;参考,碳原子的直径估计为2埃)。然后,它们将其增强版本与具有1.8埃分辨率的另一个公开的Apoferrin参考图进行了比较,并且发现两者之间的相关性得到了改善的相关性。
接下来,该团队在来自电子显微镜数据库的104个地图数据集中使用了它们的方法。对于大量比例的这些地图集,该算法改善了实验图和已知原子结构之间的相关性,并增加了细节的可见性。
作者指出,算法的明显优势在数据中揭示了数据中的重要细节,结合其易用性 - 它是一种可以在笔记本电脑处理器上执行的自动分析 - 可能会使其成为标准部分的一部分Cryo-EM工作流向前进。实际上,亚当斯已经向Phenix软件套件添加了算法的源代码,这是一个流行的自动宏分子结构解决方案,他领导开发团队。
该研究是伯克利实验室的一部分,继续努力推进冷冻电子技术的能力并开拓其对基础科学发现的用途。许多突破性发明,使得Cryo-em的发展并后来将其推向其特殊的当前分辨率,涉及伯克利实验室科学家。
参考:“通过密度修改改善了Cryo-EM地图”.Thomas C. Terwilliger,Steven J. Ludtke,Randy J.阅读,Paul D. Adams和Pavel V. Afonine,2012年8月17日,自然方法.DOI:
10.1038 / s41592-020-0914-9
郑重声明:文章仅代表原作者观点,不代表本站立场;如有侵权、违规,可直接反馈本站,我们将会作修改或删除处理。
相关阅读



猜你喜欢
-
新算法削减世界上最强大的显微镜的焦点 - 改善了Cryo-EM 3D分子结构地图
2022-05-14 -
免疫系统如何发现隐藏的入侵者的发现可能导致新的病毒和癌症治疗方法
2022-05-12 -
水如何影响自身过滤以创建高度选择性的膜,例如病毒过滤器
2022-05-12 -
新分子开发用于在化学键中存储太阳能
2022-05-09 -
量子计算的分子方法导致更少的错误
2022-05-08 -
专家讲解:如果金星上有生命,那怎么会在那里?
2022-05-07 -
麻省理工学院的化学家
2022-05-07 -
有机物是如何到达地球的?宇宙侦探痕量复合有机分子的起源
2022-05-06 -
机器学习首先确认了50个新行星– AI区分真实和“假”行星
2022-05-06 -
破裂地球的泡沫:人工智能有助于在太空中找到磁力爆发
2022-05-05 -
新阶段的纳米污水水发现 - 具有实际应用的重要基础突破
2022-05-05 -
新理论为开发量子算法提供更有效的方式提供基础
2022-05-04 -
解决超冷之谜:研究人员破解了分子消失法
2022-05-02 -
哪张脸是真实的?使用频率分析来识别“深层伪造”图像
2022-05-02 -
化学反应产生的意外涟漪违反了一份化学的中央宗旨
2022-05-02